Time: Monday 13.01.2020 13.30 – 14.00
Place: Meeting room A142, T-building
Speaker: Abbas K. Rizi
Citation: Rizi AK, Zamani M, Shirazi A, Jafari GR and Kertész J (2021) Stability of Imbalanced Triangles in Gene Regulatory Networks of Cancerous and Normal Cells. Front. Physiol. 11:573732. doi: 10.3389/fphys.2020.573732
Received: 17 June 2020; Accepted: 16 December 2020;
Published: 20 January 2021.
Balance Theory of Signed Genetic Interactions Reveals Differences in Cancerous and Healthy Cells
Abstract:
Genes are not independently functioning in the cell and their expressions are strongly correlated with each other. They communicate with each other through different regulatory effects which lead to the emergence of complex structures in the cells. Such structures are expected to be different for healthy and cancerous cells. To study the differences in the case of breast cancer, we have investigated the Gene Regulatory Network (GRN) of cells as inferred from the RNA-sequencing data using the maximum entropy principle. The GRN is a signed weighted network corresponding to the inductive or inhibitory interactions.
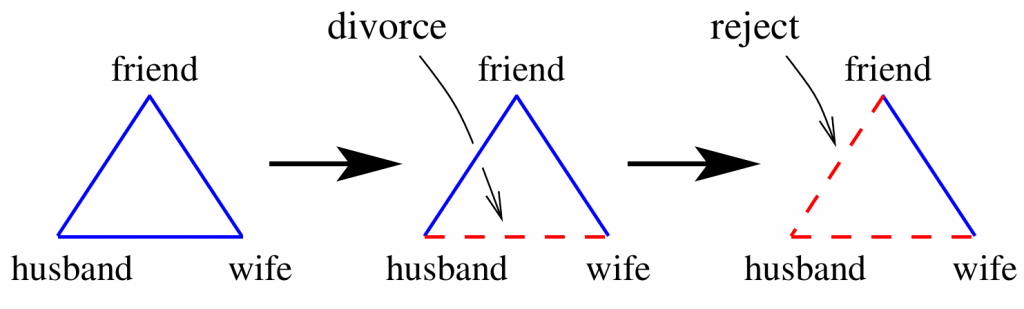
In this presentation, I will focus on a particular set of motifs in the GRN, the triangles, which can be imbalanced if the number of negative interactions in them is odd or balanced otherwise. I will show that the network in cancerous cells has fewer imbalanced triangles than in the healthy case. Moreover, in the healthy cells, imbalanced triangles are isolated from the main part of the network, while such motifs are part of the giant component of the network in cancerous cells.
- A note for the general audience: Balance Theory: From Psychology to Cancer Dynamics
- Cancer Project at CCNSD
- Click Here For the Latest Research and Reviews on GRN
Fundamental Papers on Balance Theory:
- Structural balance: a generalization of Heider’s theory, Cartwright, D., & Harary, F. (1956).
- The Energy Landscape of Social Balance, Seth A. Marvel, Steven H. Strogatz, Jon M. Kleinberg
- Dynamics of Social Balance on Networks, T. Antal, P. L. Krapivsky, S. Redner
- Social Balance on Networks: The Dynamics of Friendship and Enmity, T. Antal, P. L. Krapivsky, S. Redner
- Statistical physics of balance theory, Andres M. Belaza, Kevin Hoefman, Jan Ryckebusch, Aaron Bramson, Milan van den Heuvel, Koen Schoors
Biological Network Inference – Using the Principle of Max. Entropy
- Inferring Pairwise Interactions from Biological Data Using Maximum-Entropy Probability Models, Richard R. Stein, Debora S. Marks, Chris Sander
- Using the principle of entropy maximization to infer genetic interaction networks from gene expression patterns, Timothy R. Lezon, Jayanth R. Banavar, Marek Cieplak, Amos Maritan, Nina V. Fedoroff
- Statistical Physics of Pairwise Probability Models, Yasser Roudi, Erik Aurell, and John A. Hertz
- Inverse statistical problems: from the inverse Ising problem to data science, H. Chau Nguyen, Riccardo Zecchina, Johannes Berg
Graphical Lasso
- Regularization in Machine Learning
- Sparse inverse covariance estimation with the graphical lasso, Friedman, Jerome and Hastie, Trevor and Tibshirani, Robert (2008)
- [Slides] An Introduction to Graphical Lasso, Bo Chang (2015)
- The Graphical Lasso: New Insights and Alternatives, Rahul Mazumder, Trevor Hastie
- To obtain the estimator in programs, users could use GraphLasso() function in Python Scikit-Learn package.
- Matlab implementation of the graphical Lasso model for estimating sparse inverse covariance matrix (a.k.a. precision or concentration matrix)
Data
The data of mRNA (expression level) of 20532 genes in the case of Breast Cancer (BRCA: Breast invasive carcinoma) has been downloaded from The Cancer Genome Atlas (TCGA) project. For each gene, there exist 114 normal and 764 cancerous samples, and the expression levels have been measured with the technique of RNA sequencing (RNA-Seq). We have used the RPKM (Reads Per Kilobase transcript per Million reads.) normalized data. RPKM combines the concepts of normalizing by sample and by gene. When we calculate RPKM, we are normalizing for both the library size (the sum of each column) and the gene length. We had to reduce the number of genes because it is a difficult task to handle a 20532 by 20532 matrix computationally. For each gene, we have calculated the variance of its expression level over its samples, and finally, we have stored the first 483 genes with the highest variance due to more different activity patterns these genes show among the others. Note that there are so-called housekeeping genes that typically get transcribed continually. These genes are required for the maintenance of basic cellular function and are expressed in all cells of an organism under normal and patho-physiological conditions. Some housekeeping genes are expressed at relatively constant rates in most non-pathological situations.